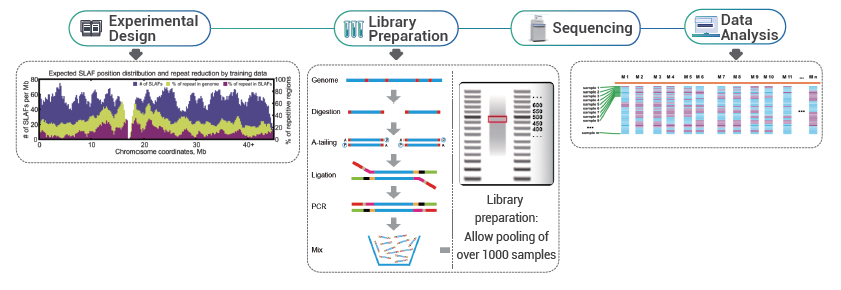
概要
ハイスループットなジェノタイピング、特に大規模集団におけるジェノタイピング解析は、遺伝的関連研究の基本ステップであり、機能的遺伝子探索や進化解析などのための遺伝的基盤を提供します。この様な研究では、全ゲノムのディープリシークエンスの代わりに、Reduced Representation Genome Sequencing(RRGS)が採用されることが多く、遺伝子マーカー探索において妥当な効率を維持しながらサンプルあたりのシークエンシングコストを最小限に抑えることができます。RRGSは、DNAを制限酵素で消化し、特定の断片サイズ範囲に焦点を当てることで、ゲノムの一部のみをシークエンシングすることでこれを実現します。様々なRRGS手法の中でも、特定遺伝子座増幅断片シークエンス(Specific-Locus Amplified Fragment Sequencing: SLAF-Seq)はカスタマイズ可能で高品質なアプローチです。BMKGene が独自に開発したこの方法は、プロジェクト毎に制限酵素セットを最適化します。これにより、反復領域を効果的に回避しながらゲノム全体に均一に分布する相当数のSLAFタグ(配列決定対象のゲノムの400~500 bps領域)が生成され、最適な遺伝子マーカーの発見が保証されます。
特長
- シークエンス条件: illumina Novaseq, PE150
- ダブルバーコードを使用したライブラリー調製により、1,000を超えるサンプルのプールが可能
- 参照ゲノムの有無により、異なるバイオインフォマティクスパイプラインを使用(有償オプション)
参照ゲノム有り:SNPおよびInDelの検出
参照ゲノム無し:サンプルのクラスタリングおよびSNPの検出
- in-silicoのプレデザイン段階で、複数の制限酵素の組み合わせをスクリーニングし、ゲノムに沿ってSLAFタグが均一に分布するものを見つけます。
- プレ実験において3つの酵素の組み合わせを3つのサンプルでテストし、9つのSLAFライブラリーを作成します。この情報に基づいてプロジェクトに最適な制限酵素の組み合わせを選択します。(fig.1)
【SLAF-Seqの特長】
- 高度な遺伝マーカー探索:ハイスループットなダブルバーコードシステムに統合することで、大規模な個体群の同時シークエンシングが可能になり、遺伝子座特異的増幅は効率を高め、タグ数が様々な研究課題の多様な要件を満たすことを保証されます。
- ゲノムへの依存度が低い: 参照ゲノムがある種にもない種にも適用できます。
- 柔軟なスキーム設計:単一酵素、二重酵素、マルチ酵素消化、および様々なタイプの酵素を、様々な研究目的や生物種に合わせて選択できます。最適な酵素設計を行うために、in-silicoによるプレ設計が行われます。
- 高効率な酵素消化:in-silicoプレ設計およびプレ実験の実施により、染色体上のSLAFタグの均等な分布(1SLAF タグ/4kb)および反復配列の減少(<5%)を備えた最適な設計を保証します。
- 豊富な専門知識: 植物、哺乳類、鳥類、昆虫、水生生物など、数百の生物種に関する5,000以上のSLAF-Seqプロジェクトを完了した実績があります。
- 独自のバイオインフォマティックワークフロー:BMKgene社は、SLAF-Seqのための統合バイオインフォマティックワークフローを開発し、最終アウトプットの信頼性と正確性を保証しています。
- 解析完了後のサポート:プロジェクト完了後 3ヶ月のアフターサービス期間を設けています。この期間中、プロジェクトのフォローアップ、トラブルシューティングの支援、結果に関する疑問やご質問に対応致します。
サービス内容
ご送付頂いた DNAサンプルを用いて、品質チェック(QCチェック)から次世代シークエンス、バイオインフォマティクス解析まで実施いたします。
【シークエンスパラメーター】
| 解析の種類 |
推奨母集団規模 |
シークエンス条件 |
| タグシークエンスの深度 |
タグ数 |
| 遺伝子地図 |
両親とその子孫(>150) |
両親: 20xWGS
子孫: 10x |
ゲノムサイズ:<400Mb
WGS 推奨
<1Gb: 100K tags
1~2Gb: 200K tags
>2Gb: 300K tags
Max 500K tags |
ゲノムワイド関連シークエンス解析(GWAS) |
≧200サンプル |
10x |
| 遺伝的進化 |
≧30サンプル
各サブグループから>10サンプル |
10x |
* 推奨シークエンス深度は、参考値です。
* QCチェックに合格したサンプルは、シークエンス量の保証対象となります。
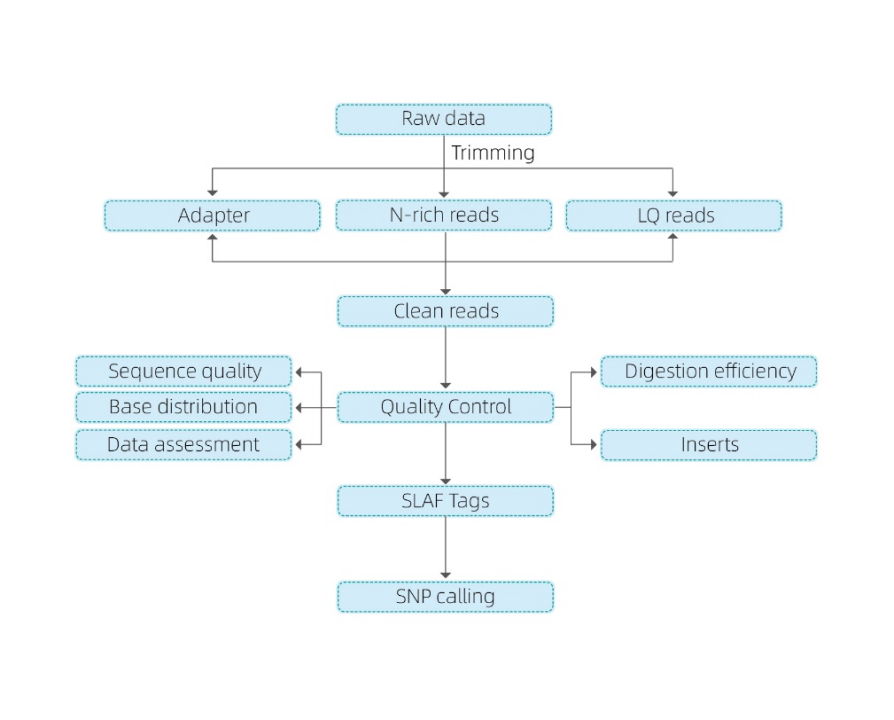
【データ解析内容】(
fig.2)
- データQC
- SLAF タグの開発
参照ゲノム有りの場合: マッピング
参照ゲノム無しの場合: クラスタリング
- SLAF タグの解析: 統計、ゲノム全体の分布
- マーカーの検出: SNP, InDel, SV および CNV コーリングとアノテーション
* パッケージの解析内容は、業務提携先のパイプラインの変更・更新等により、変更される場合があります。
作業内容
- サンプルQCチェック
- パイロット実験
- SLAF 実験
- ライブラリー作製
- シークエンス作業
- バイオインフォマティクス解析
納品物
- サンプルQCレポート
- FASTQ 形式のシークエンスデータ
- バイオインフォマティクス解析データ
- プロジェクトレポート
* USB等の記録メディアにて納品いたします。
* プロジェクトによって、納品物が変更される場合があります。
サンプル条件
| サンプルタイプ |
量(Qubit®) |
濃度 |
純度(NanoDropTM) |
gDNA |
≧160ng |
≧5ng/μL |
O.D.260/280=1.6~2.5
no or limited degradation or contamination |
* gDNA 抽出からの作業をご希望の場合は、お問合せください。
1. DNase、RNaseフリーの1.5mLまたは2mL(推奨)のチューブを使用してください。
2. Nuclease-free water または TE Buffer にて、-80℃ 以下で保存してください。
3. RNase 処理を行い、DNA が分解されていないことを確認してください。
4. UV スペクトルベースの濃度測定法では、DNA 濃度が不正確になる場合がありますので、蛍光ベースの定量法でも確認して頂くことを強く推奨します。
* UVスペクトルベースで測定した場合、上記よりも多いゲノム DNA を準備してください。
5. サンプル送付先及び注意事項につきましては、「受託解析サービスの流れ」をご参照ください。
6. サンプルの最終的な品質は、業務提携先での QCチェックの結果に従います事、予めご了承ください。